

專門作密度泛函計算的軟體
ADF量子化學軟體基於密度泛函理論(DFT),主要應用於量子化學的計算,也可廣泛應用於醫藥化學、材料學等研究及應用。本軟體特別應用於同類和異類催化、無機化學、重元素化學、生物化學及多種光譜學,包括分子ADF程式、週期結構程式BAND(專門用於計算週期性體系)和圖形用戶介面程式ADF-GUI,可在圖形介面下創建ADF計算任務並以圖形顯示結果。
使用ZORA和全電子基組有效地建模
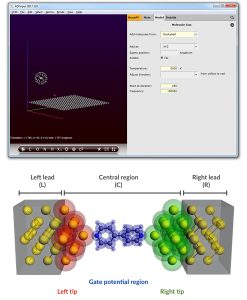
ADF通常用於研究具有重原子的過渡金屬絡合物和分子,因為周期表中的所有元素都可以使用ZORA相對論方法和全電子基組準確有效地建模。ADF提供獨特的功能來預測納米顆粒和有機電子材料的分子性質。
ADF強大的分子DFT代碼ADF用於化學和材料科學的許多領域。 ADF應用於分子性質和無機化學中特別強大。
系統需求
- 作業系統: Windows 7, Vista, XP with Service Pack 3/Linux (i686, x86-64, ia64, PowerPC)/IBM AIX/Mac OS X
- 記憶體: 256MB以上
- 儲存空間: 1GB-3GB
適用對象
1. 學術研究與大學實驗室:從事量子化學、理論化學或計算材料科學研究,需要可靠的 DFT 計算平台進行分子結構優化與電子性質分析,ADF 提供豐富的泛函與基底函數選項,滿足發表級研究需求。
2. 製藥與生技研發部門:在藥物分子設計階段需預測分子反應性、結合能與光譜特徵,ADF 的精準計算能力可加速先導化合物篩選流程,降低實驗成本。
3. 新材料與催化劑開發團隊:針對過渡金屬錯合物、重元素化合物或奈米材料進行電子結構與反應路徑模擬,ADF 對相對論效應的完整支援使其成為此領域的首選工具。
核心使用情境
- 【場景:重元素化合物研究】研究人員正在分析含鉑、金等重金屬的錯合物催化機制,傳統量子化學軟體在處理相對論效應時精度不足,導致計算結果與實驗數據出現明顯偏差。導入 ADF 後,研究團隊可啟用 ZORA(零階正則近似)相對論修正功能,精準模擬重元素的電子結構與鍵結特性,有效縮短理論與實驗之間的落差,加速論文發表進程。
- 【場景:藥物分子光譜預測與驗證】藥物研發團隊在合成新化合物後,需透過 NMR、UV-Vis 或 IR 光譜進行結構鑑定,但實驗量測耗時且成本高昂。透過 ADF 的光譜計算模組,研究人員可在合成前預測目標分子的理論光譜,與實驗結果進行比對驗證,快速排除錯誤結構假設,提升研發效率並節省試劑與儀器費用。
- 【場景:工業催化劑電子結構篩選】化工企業研發部門需從數十種候選過渡金屬催化劑中篩選出活性最佳的組合,若全部依賴實驗合成與測試,時程與成本均難以負荷。利用 ADF 進行高通量電子結構計算,可快速評估各候選材料的前線軌域能量、電荷分布與吸附能,在實驗前完成初步篩選,將實際合成測試聚焦於最具潛力的候選方案,顯著壓縮研發週期。
常見問題 FAQ
Q:ADF 量子化學軟體在台灣可以怎麼購買或取得授權?
A:ADF 為荷蘭 Software for Chemistry & Materials(SCM)公司開發的國際專業軟體,授權方案依使用規模與研究性質(學術/商業)有所不同。iQrator 提供軟體資訊諮詢與協助詢購服務,歡迎聯絡我們,協助您釐清適合的版本、授權類型與詢購流程。